







 En
En

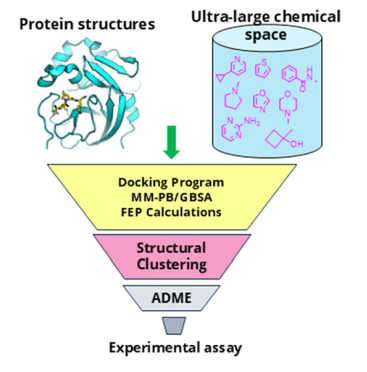
基于受体的虚拟筛选以生物大分子的三维结构为基础,通过分子对接的方法,挑选出结合模式合理、打分较高的化合物,用于后续的生物活性测试。

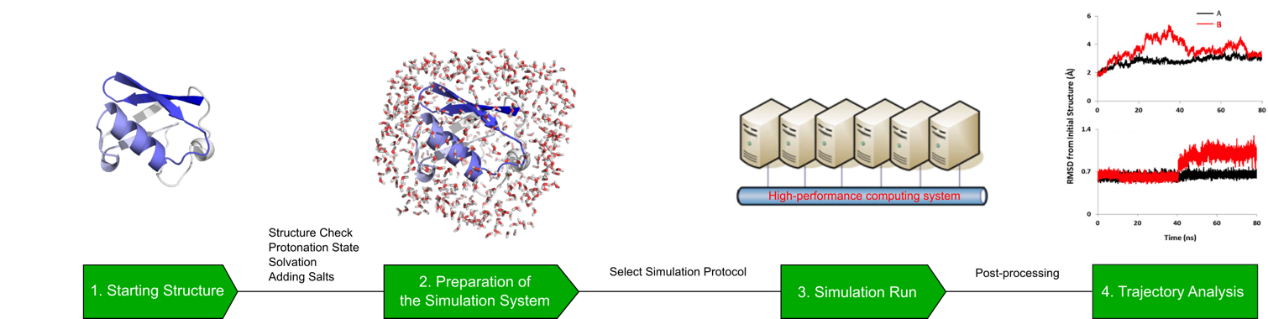
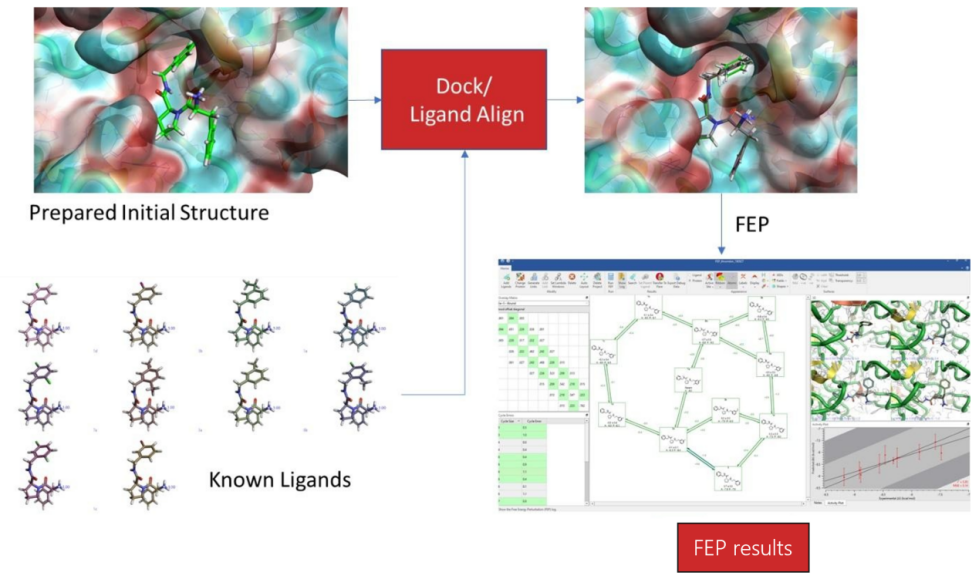
计算机辅助药物设计(Computer-aided drug design,CADD)是⼀类借助计算方法来加速药物研发的技术。平台配置有高性能计算集群以及专业的药物计算软件,可在小分子先导化合物的发现与优化阶段提供计算支持。主要提供虚拟筛选、分子对接、分子动力学模拟、结合自由能计算等服务,并持续开发及扩展更多计算服务。
 管理员:刘慧敏
管理员:刘慧敏
 联系电话:83950108,83950107
联系电话:83950108,83950107
 电子邮箱:liuhuimin@cimrbj.ac.cn
电子邮箱:liuhuimin@cimrbj.ac.cn
 放置地点:首都医科大学南校区科研楼南楼负一层
放置地点:首都医科大学南校区科研楼南楼负一层

基于受体的虚拟筛选以生物大分子的三维结构为基础,通过分子对接的方法,挑选出结合模式合理、打分较高的化合物,用于后续的生物活性测试。
基于配体的虚拟筛选即基于药效团模型的虚拟筛选,是根据现有药物的结构、理化性质和活性关系的分析,建立定量构效关系或药效团模型,从而筛选具有潜在活性的化合物。
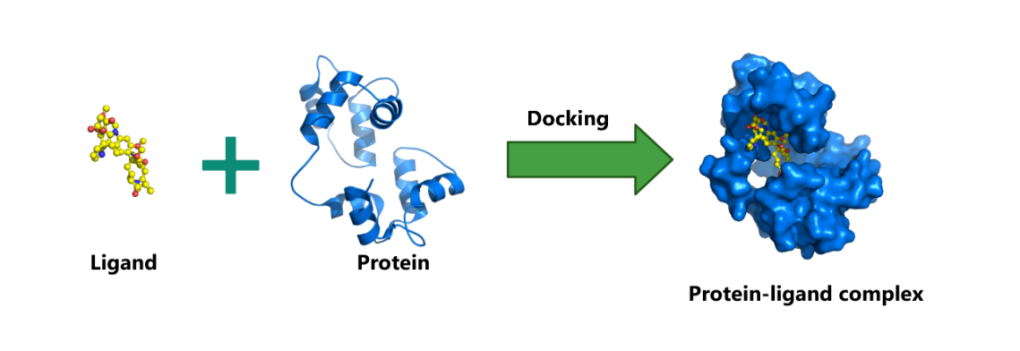
刚体对接:在对接过程中,研究体系的构象不发生变化。适合考察比较大的体系,如蛋白质和蛋白质之间以及蛋白质与核酸之间的对接。
半柔性对接:对接过程中,研究体系尤其是配体的构象允许在一定的范围内变化。适合处理大分子和小分子间对接,对接过程中,小分子的构象一般是可以变化的,但大分子是刚性的。
柔性对接:对接过程中,研究体系的构象可以自由变化。一般用于精确计算分子间的相互作用情况,由于计算过程中体系的构象可以变化,因此计算量最大。



 北京市丰台区右安门外西头条10号
北京市丰台区右安门外西头条10号
 010-86738999
010-86738999
 cimr@cimrbj.ac.cn
cimr@cimrbj.ac.cn
